Adverse events in clinical trials
During any clinical trial, safety is top of mind. This is assured through a series of checks that keep sponsors and investigators accountable and in compliance with FDA requirements for investigational new drug (IND) safety reporting. As with any IND clinical trial, adverse events may occur.
An adverse event (AE) is any sort of contrary medical experience associated with human use of the drug, whether or not the experience may be drug related. Any AE connected with drug use sets off a complex process for sponsors and regulators. There are multiple questions to consider in terms of reporting under 21 CFR 312.32 (c). Since this can be a daunting procedure, the Bracken regulatory team has included in this post a user-friendly flowchart that efficiently guides sponsors through the steps of reporting.
Questions to consider when an AE occurs
To assure understanding and remain in compliance, the FDA offers specific definitions for events that may occur in an IND clinical trial. Depending on the seriousness of the AE, the FDA may need to be notified in writing and in an expedited timeframe. It is particularly important to be mindful of AEs that are suspected, unexpected, and serious.
An AE is considered suspected when there is reason to believe the drug caused the event. For this to occur, there must be evidence linking the drug to the AE.
An AE is considered unexpected when the reaction experienced is not listed in the investigator’s brochure or the reaction is more severe than accounted for. If the language in the brochure is not specific enough to determine if the reaction is unexpected or if there is no brochure available/required, this may also meet the unexpected standard.
An AE is considered serious if it leads to any of the following: death, a life-threatening event, hospitalization or extension of an existing hospitalization, a significant or chronic inability to perform normal functions of daily life, or a birth defect or other congenial abnormality.
When to notify the FDA
After reviewing all information, if the event is determined to have a suspected unexpected serious adverse reaction (SUSAR), the sponsor must follow a 7-day or 15-day timeline to notify the FDA in writing of the event, depending on the severity of the reaction.
A life-threatening reaction puts the individual at risk of death immediately. A reaction may also be considered serious if it compromises the individual and requires medical attention to prevent one of the events considered serious from occurring.
If the SUSAR is determined to be life-threatening and/or results in death, the report must be submitted within 7 calendar days. A SUSAR that does not result in death, nor is found to be life threatening, must be submitted within 15 calendar days.
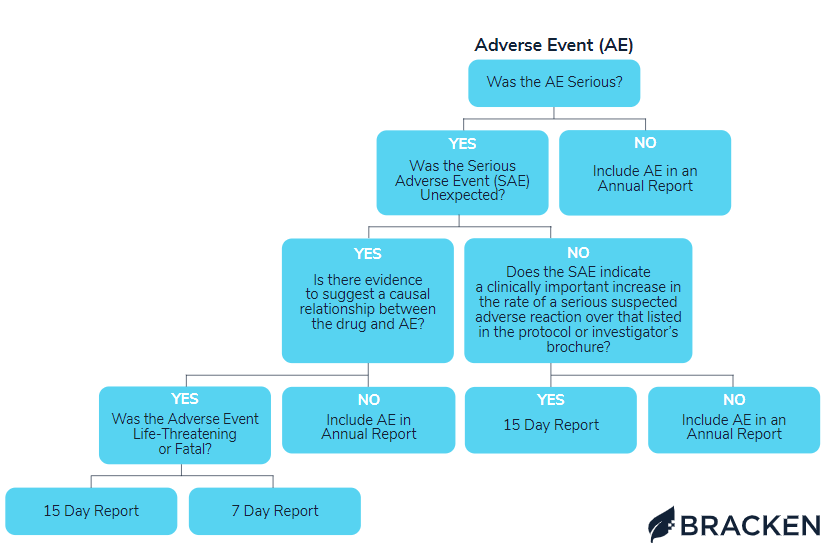
AE reporting visual flowchart
To clarify what AEs must be reported and when, please refer to the comprehensive flowchart below. The Bracken regulatory team designed this visual guide to address each possible event occurrence and direct how to proceed.
How Bracken’s regulatory expertise can help
For further clarification on AEs, SUSARs, and the regulatory process in general, Bracken’s regulatory team is here to offer guidance and solutions.



