Why It Matters
If you’re in the business of developing drugs for the treatment or prevention of diseases, you will need the review and approval of the regional regulatory agency to market your product within its jurisdiction. These groups regulate the safety and quality of drugs prior to and after marketing approval. But regulatory bodies vary widely across the world. Knowing how the relevant agency operates could save you vast amounts of time and money and help ensure that your product reaches the market according to plan. From a global perspective, a thorough understanding of the various regulatory authorities can help shape your product development and launch plans; including harmonizing your strategy to fulfill global regulatory requirements.
Start Using TrialTrends: A Free Tool That Visualizes Clinical Trial Registration Rates
The Role of Regulatory Agencies
Pharmaceutical regulations play an important role in pharmaceutical product oversight by helping assure product quality and the rigor of pre-clinical and clinical data to support claims of safety and efficacy for approved drugs. They may also require that new drugs provide additional advantages to patients over existing medicines, such as improved formulation, potency, dosing regimens, or delivery routes. While some countries, such as the US, have established a single agency to oversee these regulatory functions, others, such as the Europe operate on a regional level across multiple countries.
Though these agencies differ in how they evaluate safety and efficacy, and thus what they allow to enter the market in their respective regions, regulators generally all work to ensure that:
-
Medicines meet clear standards of quality, safety, and efficacy prior to approval,
-
Approved medicines are manufactured, stored, distributed, and dispensed according to strict guidelines, and
-
Promotion and advertising are not misleading, biased, or outside the limits of the approved indication
How They Differ
A recent study of regulators in established pharmaceutical markets (including the United States, Europe, Japan and China) reported that the US Food and Drug Administration (FDA) has approximately 2,000 internal reviewers while the European Medicines Agency (EMA) relies on a network of over 4,500 external reviewers who provide the agency with scientific and regulatory expertise. By contrast, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) operates with about 560 technical reviewers, and China’s National Medical Products Administration (NMPA) had only about 120 staff in its Center of Drug Evaluation as of 2015.
But the differences don’t end with the number of reviewers. Marketing authorization applications (MAAs) submissions and approvals also vary by region and regulatory agency. While the FDA received 35 MAA submissions (termed New Drug Application [NDA] in US) in 2015, the EMA received 61, and Japan 127. MAA approvals for new therapeutics in 2016 were 22 for the FDA, 27 for EMA, and 48 for PMDA. Regulatory fees also vary widely by region: while the fee to review an MAA costs approximately $1,000 USD in India (~50,000 rupees), it costs about $2.6M USD here at home. Perhaps most significant, new drug approval times also vary by agency, ranging from as short as 1 month to several years.
These differences will undoubtedly play a significant role in developing an effective product launch strategy, where timing is everything. Many pharmaceutical product development teams start conducting market research and developing their launch plan as far back as two years prior to NDA approval. Ideally the indication and the market research should be conducted as part of the product development path forward. Knowing how the different agencies work, and what their requirements, timelines, and costs are, will undoubtedly help you develop a cogent plan.
The Regulatory Bodies You Should Know
In Table 1 below, you’ll find pertinent information about 7 regulatory authorities you’ll want to consider in your drug development and product launch plans.
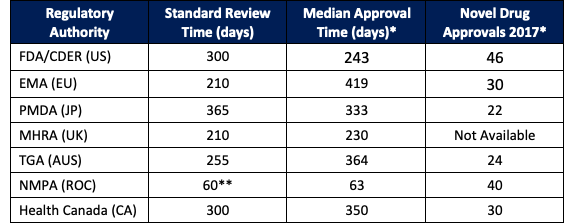
Click on the agency name to find contact information for each authority:
*Data is only available through 2017.
**China’s standard review time moved from and average 4.49 years in 2015 to 53 days in 2018, due largely to foreign data being accepted for registration if there is no ethnic difference in the patient population.
In addition to the links above, you may find it useful to access information on:
-
The FDA Center for Biologics and Research - CBER is the agency charged with protecting the public health “through the regulation of biological and related products including blood, vaccines, allergenics, tissues, and cellular and gene therapies”
-
The FDA Center for Devices and Radiological Health - CDRH is responsible for regulating the manufacture, repackaging, relabeling, and/or import of medical devices sold in the United States. CDRH also regulates radiation-emitting electronic products such as lasers, x-ray systems, and ultrasound equipment.”
-
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use - ICH focuses on the scientific and technical aspects of drug registration. Its mission is to achieve greater harmonisation worldwide to ensure that safe, effective, and high quality medicines are developed and registered in the most resource-efficient manner.The ICH has evolved in response to the increasingly global face of drug development.
-
ClinicalTrials.gov is a comprehensive database of clinical studies conducted around the world.


